Nouvelles médicales

Transplantation de cellules souches hématopoïetiques Autologues
Ces 20 dernières années, la transplantation de cellules souches hématopoïétiques autologues a été utilisée épisodiquement dans certaines formes graves de SEP. Il y a peu, ce traitement a bénéficié d'un regain d'attention. Une émission de Koppen (programme de la VRT) a soulevé de nombreuses questions du côté des patients. Dans les lignes qui suivent, nous nous penchons sur une récente publication qui fait le point sur ces 20 années d'expérience dans cette thérapie.
Introduction
La transplantation de cellules souches hématopoïétiques s'utilise de manière classique dans les cas de cancer du sang. Le traitement repose sur la destruction des cellules cancéreuses au moyen d'une chimiothérapie hautement dosée, après quoi le système immunitaire est relancé grâce à transplantation de cellules souches autologues ou allogéniques. Dans le cas d'une transplantation allogénique, le donneur et le receveur diffèrent au niveau immunologique et les cellules souches transplantées peuvent donner naissance à un nouveau système immunitaire mais aussi détruire les cellules cancéreuses qui auraient survécu à la chimiothérapie. Cet effet secondaire positif, la suppression des cellules tumorales, porte le nom de réaction GVT (graft versus tumor, ou greffe contre tumeur). Malheureusement, les cellules transplantées risquent également de viser les cellules du receveur, donnant lieu des réactions potentiellement létales. C'est ce qu'on appelle l'effet greffe contre hôte. Compte tenu de cette éventualité, la transplantation de cellules souches hématopoïétiques autologues est privilégiée dans le cas de maladies auto-immunes comme la SEP, car ces cellules autologues transplantées renouvellent ou relancent le système immunitaire sans ce risque d'effet greffe contre hôte parfois fatal.
Procédure
Plusieurs protocoles sont appliqués, tant pour la préparation de la transplantation (chimiothérapie) que pour la transplantation proprement dite. Par contre, le nombre total de patients traités reste relativement faible. Nous allons donc tenter de nous concentrer sur les avantages et les inconvénients observés dans les différents programmes thérapeutiques.
Résultats en termes d'évolution clinique
Toutes les études de phase I/II publiées montrent que la transplantation de cellules souches hématopoïétiques autologues permet une stabilisation, voire une amélioration de la maladie, du moins à moyen terme. Une analyse rétrospective du Group for Blood and Marrow Transplantation (EBMT, Groupe européen pour la transfusion sanguine et la greffe de moelle osseuse) indique qu'après 3 ans, 74 % des patients étaient restés stables, ce chiffre passant à 45 % après 5 ans. Environ la moitié des personnes traitées constaterait une amélioration de leur EDSS, une mesure de la répercussion fonctionnelle de la maladie. Un effet prononcé s'observe au niveau des flambées, avec un contrôle presque total des attaques les premières années qui suivent la thérapie.
Résultats en termes d'évolution radiologique
La plupart des études font état d'un effet important sur le nombre de lésions captant le produit de contraste, mais aussi sur l'apparition de nouvelles lésions. Par contre, il semble que le traitement n'ait aucune influence sur le développement d'une atrophie cérébrale ou la diminution progressive du volume cérébral à mesure que la maladie évolue.
Sécurité
La morbidité associée à la transplantation, soit le risque de décéder des suites de complications dues à cette procédure, s'élève aujourd'hui à 1 ou 2 %. Ce taux était encore de 7,3 % pour la période 1995-2000. Ce recul du risque de mortalité est vraisemblablement lié à des adaptations des protocoles, à une meilleure sélection des patients ainsi qu'à une plus grande expérience de la part des hématologues et des neurologues.
Parmi les autres conséquences courantes du traitement, citons la fièvre due à un déficit en globules blancs, les infections et la diarrhée. L'apparition d'autres maladies auto-immunes, touchant notamment la thyroïde, compte au nombre des complications tardives. Enfin, le traitement peut également provoquer l'infertilité.
Conclusion
Après 20 années d'utilisation, la transplantation de cellules souches hématopoïétiques autologues dans le cadre de la SEP a évolué jusqu'à devenir une option de traitement envisageable qui s'accompagne cependant d'un risque de mortalité de 1 à 2 % et d'effets secondaires graves et permanents. À l'heure actuelle, elle est donc considérée comme une option à prendre en compte pour un groupe de patients soigneusement sélectionnés. Ceux-ci doivent être atteints d'une SEP débutante (maladie ayant débuté depuis moins de 5 ans, patient âgé au maximum de 40 ans) mais extrêmement agressive, dont les poussées se poursuivent malgré un traitement « classique ». À l'heure actuelle, d'autres options de traitement sont également accessibles à ce groupe de patients, dont le Tysabri et le Lembrada. La place précise de chacune de ces thérapies n'est pas encore claire, et des études comparatives s'imposent pour s'en faire une idée plus nette.
- Pr. Bénédicte Dubois
- Service Neurologie Universitaire Ziekenhuizen Leuven - KU Leuven
Source :
- D. Curro et al.
Autologous hematopoietic stem cell transplantation in multiple sclerosis: 20 years of experience
Neurol. Sci. 2016, Apr. 12
L'influence du stress sur la sclérose en plaques
Introduction
La SEP est une maladie inflammatoire du système nerveux central dont l'évolution est imprévisible. Pendant les premières années de la maladie la plupart des personnes passent par des poussées avec des plaintes neurologiques nouvelles ou aggravées. Les poussées correspondent à l'apparition de nouveaux foyers d'inflammation à hauteur de la myéline, visibles sur les images par résonance magnétique du cerveau et de la moelle épinière. En outre de nombreuses personnes atteintes de SEP déclinent progressivement, ce qui correspond à une dégradation grandissante à hauteur des fibres nerveuses. Ce processus de dégradation grandissante n'est pas ou seulement très tardivement visible sur les images par résonance magnétique du système nerveux central.
L'évolution de la SEP diffère de personne à personne. Nous ne savons pas pourquoi. Peut-être est-ce dû à une combinaison de facteurs génétiques et environnementaux.
Cellules-souches: remyélinisation et sclérose en plaques
- Définition des cellules-souches : une cellule-souche est définie comme une cellule ayant la capacité de se renouveler elle-même d'une part, de générer des types cellulaires très spécialisés d'autre part. Les cellules-souches isolées à partir de stades précoces de développement possèdent des potentialités plus élevées de se différencier en de multiples cellules différentes que celles prélevées à un stade plus tardif. Ce sont donc les cellules-souches embryonnaires qui sont les plus « pluri-potentes », c'est-à-dire qui ont la plus grande potentialité en culture cellulaire de générer différents types de cellules, en particulier des cellules nerveuses et des cellules gliales, dont l'oligodendrocyte qui fabrique la gaine de myéline.Outre les cellules-souches embryonnaires, il existe des cellules-souches fœtales, et des cellules-souches adultes, provenant de la moelle osseuse (cellules-souches mésenchymateuses), des cellules-souches hématopoiétiques (précurseurs des cellules sanguines) et des cellules-souches neurales (situées dans la profondeur du cerveau, le long de la paroi des ventricules contenant le liquide céphalorachidien). Il n'est pas techniquement si facile de provoquer la différenciation de ces différents types de cellules-souches en une cellule cible bien différenciée, par exemple en une cellule nerveuse ou en un oligodendrocyte.Par ailleurs, sont présentes de manière diffuse dans le système nerveux central (cerveau et moelle épinière) des « cellules précurseurs des oligodendrocytes » qui peuvent être aussi considérées comme des cellules-souches spécifiques de ce type cellulaire. Ces cellules précurseurs peuvent partiellement mais efficacement remyéliniser certaines plaques de sclérose en plaques. La gaine de myéline reformée par ces cellules précurseurs est généralement plus fine, moins épaisse, et d'une longueur plus limitée. Malheureusement, cette myéline ainsi reformée peut être à nouveau la cible du processus destructeur et être donc détruite.
- La neuroprotection dans la sclérose en plaques : il s'agit là d'un objectif thérapeutique majeur qui n'est pas encore rencontré par nos thérapeutiques actuelles. Cette neuroprotection a deux composantes : 1°) la prévention de la perte des fibres nerveuses appelées axones, soit par transsection durant l'inflammation aiguë, soit chroniquement, suite à la perte de leur gaine de myéline protectrice, au cours des mois et des années ; 2°) la promotion de la remyélinisation dans les endroits où la gaine de myéline a été détruite. Ces deux phénomènes sont liés car il est évidemment impossible de remyéliniser les axones si ceux-ci sont en état de dégénérescence. On a de bonnes raisons de penser que la phase progressive de la maladie, observée en moyenne 10 à 15 ans après la première poussée, est liée directement à cette dégénérescence des axones. Par ailleurs, 15% des patients atteints de sclérose en plaques n'ont pas de poussée ni de rémission et commencent d'emblée leur maladie dans une phase progressive de dégénérescence axonale.
- Thérapeutique par cellules-souches dans la sclérose en plaques : on peut imaginer deux mécanismes, qui d'ailleurs ne s'excluent pas mutuellement : soit remplacer les cellules-souches, et en particulier les cellules précurseurs des oligodendrocytes par des cellules provenant d'une source externe, injectées au patient ; soit, augmenter la réparation endogène en utilisant les cellules-souches de la personne elle-même et en particulier en stimulant les cellules précurseurs des oligodendrocytes.
a) la réparation cellulaire par des cellules exogènes pose de nombreux problèmes. Il faut en effet que les cellules-souches se différencient en une quantité importante du type cellulaire souhaité, par exemple la cellule précurseur de l'oligodendrocyte, que cette différentiation soit stable, qu'il n'y ait pas de surproduction cellulaire évoluant vers une tumeur ; et enfin que les cellules-souches se dirigent vers la zone pathologique. On sait cependant que les cellules-souches ont une attraction innée vers les zones pathologiques nécessitant une réparation, cette propriété étant appelée « pathotropisme ».
b) la promotion des mécanismes de réparation endogène : il s'agit d'une approche sans doute plus réaliste et plus faisable dans le futur proche. On sait que dans les plaques de sclérose en plaques on trouve fréquemment des cellules précurseurs de l'oligodendrocyte mais elles semblent inhibées par l'environnement inflammatoire, ce qui empêche la remyélinisation. De nombreuses études essaient actuellement d'élucider les mécanismes pro- et anti-remyélinisants, et les molécules favorisant ou bloquant cette remyélinisation. C'est ainsi qu'une de ces molécules, appelée lingo, est la cible d'un anticorps monoclonal anti-lingo qui favorise la remyélinisation dans des modèles expérimentaux.
Les premiers essais en clinique humaine ont démarré dans le cadre de la névrite optique comme premier symptôme de sclérose en plaques. Il s'agit de savoir si cet anticorps anti-lingo permet de protéger le nerf optique, d'empêcher la dégénérescence des fibres nerveuses et ainsi de maintenir une meilleur acuité visuelle. De même, une autre étude de faisabilité a déjà été réalisée en utilisant des cellules mésenchymateuses prélevées chez les patients eux-mêmes pour traiter des formes secondaires progressives de sclérose en plaques. Les résultats initiaux étaient encourageants pour la fonction visuelle.
Conclusion
L'étude des cellules-souches dans le traitement de la sclérose en plaques nous apportera certainement des indications précieuses sur les mécanismes de la maladie et sur la question clé de la neuroprotection.
Les problèmes éthiques (utilisation de cellules-souches embryonnaires ou fœtales) et techniques restent extrêmement importants cependant. Les cellules-souches provenant du sujet lui-même ne posent pas de problèmes éthiques, ne sont pas détruites par le système immunitaire de la personne concernée, mais ont moins de potentialité de se différencier dans le type cellulaire souhaité. Même dans le cerveau d'un homme adulte, il existe des cellules précurseurs ayant toujours des capacités de se différencier et de remplacer d'autres cellules détruites par un processus pathologique.
Professeur Christian SINDIC
Juillet 2013
Neuromyélite optique : une variante de la SEP ?
La neuromyélite optique (NMO ou maladie de Devic) est une maladie inflammatoire qui touche principalement les nerfs optiques et la moelle épinière. Elle se caractérise également par une nette prédominance chez les femmes, la présence de longues lésions dans la moelle épinière à l’IRM et l’absence de bandes oligoclonales dans le liquide céphalorachidien. Certains symptômes ressemblent donc à ceux de la SEP, d’autres diffèrent, et on s’est longtemps demandé s’il existait un lien précis entre les deux maladies. Des études ont toutefois permis d’identifier une protéine directement liée à la NMO : les anticorps dirigés contre l’aquaporine 4. Pourtant, on retrouve aussi cette protéine chez certains patients souffrant fréquemment d’une inflammation de la moelle épinière ou des nerfs optiques, mais pas la combinaison des deux. Il s’agit donc d’un spectre de maladies NMO qui sont apparentées et dont les symptômes ressemblent à première vue à ceux de la SEP, mais finalement ils en diffèrent. Cet article présente quelques aspects importants des maladies NMO.
Manifestation générale
La NMO se manifeste généralement à partir de 40 ans, bien qu’un premier épisode soit possible à tout âge. Elle est plus fréquente chez les femmes que chez les hommes. Parmi les personnes touchées par la NMO, 3 % ont un membre de leur famille qui en souffre également, ce qui laisse supposer qu’il n’y a pas de cause héréditaire, mais bien une prédisposition génétique. On n’en sait pas plus à ce sujet pour l’instant.
Caractéristiques cliniques
La plupart des patients présentent un parcours de la maladie qui se caractérise par des poussées. Ces attaques sont généralement plus sévères que celles décrites dans la SEP et provoquent une baisse de la vision (suite à l’inflammation des nerfs optiques), ainsi qu’une perte de sensibilité, une faiblesse musculaire et des troubles de la vessie (suite à l’inflammation de la moelle épinière). Une évolution progressive secondaire, au cours de laquelle les plaintes augmentent progressivement, mais sans rechutes, est rare dans la NMO, contrairement à la SEP. La fatigue est une plainte récurrente, tant chez les patients souffrant de SEP que de NMO.
Caractéristiques radiologiques
Si une IRM (imagerie par résonance magnétique) cérébrale présente souvent clairement plusieurs lésions de la substance blanche, c’est nettement moins le cas dans la NMO. L’IRM cérébrale peut même être normale. En revanche, l’IRM médullaire révèle souvent de longues lésions inflammatoires.
Test sanguin
Comme en témoignent les caractéristiques mentionnées ci-dessus, il n’est pas toujours évident de faire la distinction entre la SEP et une maladie du spectre NMO. Par bonheur, des études scientifiques ont abouti en 2004 à l’identification d’anticorps dirigés contre l’aquaporine 4 qui sont détectés chez les patients atteints de NMO, mais pas chez ceux qui souffrent de SEP classique. Dans la NMO, d’autres maladies auto-immunes sont également plus fréquentes, comme une affection thyroïdienne auto-immune, un lupus, une myasthénie auto-immune, etc. L’examen sanguin fait également état d’un nombre plus important d’auto-anticorps dans la MNO, même sans maladie auto-immune spécifique.
Traitement
A l’instar de la SEP, le traitement de la MNO repose sur différents principes thérapeutiques : le traitement d’une phase aiguë, la prévention des récidives, le traitement des symptômes et la revalidation. Ces deux derniers ne différent pas du traitement de la SEP.
Une phase aiguë est traitée à l’aide de cortisone administrée au patient par voie intraveineuse, comme dans la SEP. Ensuite, la cortisone est prise par voie orale pendant quelque temps. Si la poussée persiste, le patient est soumis à un traitement par plasmaphérèse qui permet de soustraire du sang les substances toxiques, comme les anticorps dirigés contre l’aquaporine 4.
La prévention des poussées est spécialisée. L’important est de savoir qu’un traitement de référence dans la SEP, comme les interférons ou l’acétate de glatiramer, ne constitue pas un bon choix en cas de NMO.
Conclusion
Il est évident que des similitudes existent entre la SEP et la NMO au niveau des symptômes, de l’évolution des poussées et du traitement de ces épisodes. C’est la raison pour laquelle cette affection a naturellement sa place dans un site destiné aux patients atteints de SEP. Ces deux maladies présentent toutefois des différences intrinsèques qui se manifestent par les différentes caractéristiques détectées à l’IRM, la présence d’anticorps dirigés contre l’aquaporine 4 et un traitement d’entretien différent.
Prof. B. DUBOIS
Service de neurologie - Universitaire Ziekenhuizen Leuven - KU Leuven
Avril 2013
Source
JACOB, A., et al., Current concepts of neuromyelitis optica (NMO) and NMO spectrum disorders,
Journal of Neurology, Neurosurgery and Psychiatry 2012, en cours d’impression.
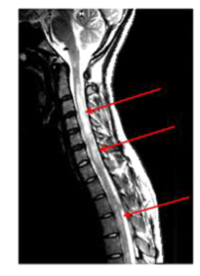
IRM en cas de maladie de Devic. Les flèches rouges signalent l’inflammation.
La neuromyélite optique (Neuromyelitis Optica Spectrum Disorder : NMOSD) est une affection démyélinisante inflammatoire du système nerveux central qui diffère nettement de la sclérose en plaques (SEP). Néanmoins, ces deux maladies partagent des points communs historiques et des mécanismes dysimmunitaires.
La fréquence du NMOSD est rare dans les pays occidentaux. Cette affection est plus fréquente en Asie (Japon). Les données épidémiologiques actuelles permettent de considérer que le NMOSD est observé avec une incidence de 1 % des patients SEP (soit 150 patients environ en Belgique). Pendant de nombreuses années, la maladie a été considérée comme une forme particulière de SEP, responsable d’une atteinte préférentielle des voies optiques et de la moelle épinière. Le pronostic fonctionnel était assez péjoratif, avec des patients qui présentaient rapidement des troubles visuels et moteurs sévères dans les cinq années qui suivaient les premières manifestations. Initialement appelée maladie de Devic, elle a été longtemps traitée comme une SEP. Contrairement à la SEP, le handicap observé chez des patients qui souffrent de NMOSD est la conséquence des poussées qui peuvent être très aigües. Il n’y a pas de formes progressives de NMOSD.
Les progrès de l’IRM et de l’immunologie ont complètement changé le cadre de cette affection. L’IRM a permis de détecter des lésions différentes de celles initialement décrites (atteinte cérébrale supra-tentorielle, atteinte du tronc cérébral et de la région périaqueducale).
En 2004, une importante publication a permis de démontrer la présence d’anticorps spécifiques dirigés contre un canal à l’eau (AQP4Ab), situé sur la membrane des astrocytes. Les astrocytes sont des cellules très importantes du système nerveux central (SNC) qui sont indispensables à la fonction des neurones. Ils sont une sorte de « pont fonctionnel » entre les vaisseaux sanguins et les autres cellules (neurones et oligodendocytes). Les anticorps anti-AQP4 sont des IgG1 fabriqués par des cellules immunitaires (plasmocytes dérivés des lymphocytes B). Ils sont présents chez environ 80 % des patients adultes qui souffrent de NMOSD. L’activation de ces lymphocytes B et leur évolution vers des plasmocytes qui synthétisent les AQP4Ab dépend d’une stimulation par des lymphocytes T de la famille des Th17 (T helper). Les anticorps sont produits dans le sang périphérique et franchissent la barrière hémato-encéphalique. Ils se fixent sur les pieds astrocytaires (prolongement des astrocytes vers les vaisseaux sanguins capillaires). Cela provoque une activation du système du complément. Ce dernier est un ensemble de molécules qui participent à la réaction inflammatoire et à ses conséquences toxiques pour les cellules. La réaction inflammatoire est donc liée à l’activation du complément (CDC) et des anticorps (ADCC). Les astrocytes sont le siège d’un œdème et une rupture de la barrière hémato-encéphalique se met en place. La souffrance des astrocytes provoque un dysfonctionnement des neurones et des oligodendrocytes. Cet ensemble de phénomènes permet l’arrivée massive d’autres globules blancs (polynucléaires neutrophiles et éosinophiles) qui aboutissent à une inflammation très importante du tissu. Les neurones sont rapidement soumis à un stress cellulaire majeur, ne fonctionnent plus et meurent. Cette cascade inflammatoire est donc très rapide et aboutit aux symptômes que le patient ressent. Le rôle pathogène (responsable de la maladie) des AQP4Ab a été démontré par le déclenchement d’un NMOSD chez des animaux de laboratoire à qui on a administré du sérum de malades atteints.
Un autre anticorps dirigé contre une protéine de la myéline (MOG) a été associé au NMOSD. Son rôle exact est moins clair. Il n’est retrouvé que chez 4 à 11 % des patients qui présentent ce tableau clinique, plus souvent chez des sujets plus jeunes, voire des enfants. Seule la persistance de ces anticorps anti-MOG est un élément qui conduit à l’instauration d’un traitement spécifique. Les analyses microscopiques du tissu atteint ont montré des anomalies réversibles de la myéline, sans activation du complément et sans infiltration inflammatoire importante.
Enfin, dans 14 à 22 % des cas de NMOSD, aucun anticorps anti-AQP4 ou anti-MOG n’est mis en évidence et les critères de diagnostic sont plus stricts avant de considérer qu’il s’agit bien de cette maladie.
Les critères de diagnostic du NMOSD ont été clarifiés et publiés en 2015 avec des conséquences importantes dans la prise en charge thérapeutique des patients. En effet, en présence d’une seule manifestation typique de la maladie, la démonstration d’un AQP4Ab dans le sang, permet de poser le diagnostic et d’instaurer un traitement de fond efficace destiné à éviter la survenue de rechutes. Ces dernières peuvent être sévères et associées à un handicap résiduel important. La prise en charge des patients dès le stade précoce est donc fondamentale.
La stratégie thérapeutique
Les poussées aigües de NMOSD
Les poussées peuvent être responsable d’un handicap important et le traitement précoce des rechutes est un élément fondamental. Habituellement, la première phase de traitement d’une poussée consiste en l’administration intraveineuse d’une haute dose de cortisone, comme cela est le cas dans la sclérose en plaques. Le patient va recevoir 1000 mg de Solumedrol par voie intraveineuse, par jour, pendant 3 à 5 jours consécutifs. Le délai de prise en charge doit être le plus court possible, dès que la rechute est identifiée. L’évolution du tableau déficitaire sous traitement est généralement rapide. Si une amélioration significative n’est pas obtenue après quelques jours, des échanges plasmatiques (PLEX ou plasmaphérèses) sont recommandés. Les PLEX sont une technique qui permet d’extraire les anticorps anormaux du sang à l’aide d’une filtration qui est très proche de ce qui est effectuée au cours d’une dialyse. Cela sous-entend que la mise en place d’un cathéter veineux central de gros calibre doit être effectuée, du moins transitoirement. Le patient est alors raccordé à une machine qui filtre le sang. Ce dernier passe sur une membrane qui retient les anticorps. Le sang est ensuite réinjecté au patient lui-même. Les PLEX sont généralement réalisés à raison de 5 à 7 séances sur une période de 2 semaines. Contrairement à la pratique dans le domaine de la SEP, le traitement par corticoïdes et/ou PLEX doit être suivi d’une période de corticothérapie par voie orale pendant quelques semaines. Certains auteurs recommandent même l’utilisation d’une immunosuppresseur puissant dans les suites immédiates des PLEX pour éviter tout phénomène de rebond. Jusqu’à l’heure actuelle, aucune donnée scientifique ne permet de prédire la réponse au traitement par corticoïdes ou PLEX, en particulier en fonction de la présence des anti-AQP4Ab. Certaines études observationnelles semblent montrer que les patients qui présentent un NMOSD associé à des anticorps anti-MOG ont une réponse particulièrement favorable aux corticoïdes mais avec un risque relativement élevé de rebond à l’arrêt du traitement.
Les traitements « classiques » de maintenance
Le Rituximab
Il s’agit d’un anticorps monoclonal cytotoxique chimérique dirigé contre le récepteur CD20. Un anticorps monoclonal est fabriqué par des cellules qui synthétisent systématiquement la même molécule qui reconnaît sa cible avec une précision parfaite. Lorsque l’anticorps se fixe sur son récepteur, il induit une cascade d’évènements qui va provoquer la destruction de la cellule. Ce médicament a été initialement développé et approuvé dans le traitement de tumeurs lymphatiques (lymphomes). Les cellules qui portent la carte d’identité CD20 sont essentiellement des lymphocytes B. Une petite proportion de lymphocytes T sont également porteurs de cet identifiant. Le terme chimérique signifie qu’une partie de l’anticorps comporte des séquences d’acides aminés qui viennent de la souris. Après perfusion de cet anticorps monoclonal, la molécule persistent dans l’organisme pour une période de 120 heures à 6 mois. Plusieurs schémas posologiques sont recommandés mais la plupart des centres utilisent une dose initiale de 1 gr répétée après 15 jours ou 375 mg/m2 toutes les semaines pendant 4 semaines. Le médicament va détruire tous les lymphocytes B après environ 1 mois. Il faut cependant signaler que seule une très petite proportion (1%) du médicament franchit la barrière hémato-encéphalique et pénètre dans le liquide céphalo-rachidien. L’efficacité du Rituximab n’a pas été étudiée dans des essais internationaux classiques. Les données actuelles reposent sur des séries publiées et des analyses post-hoc. L’effet favorable du médicament est important, de l’ordre de 80 % de réduction de la fréquence des poussées, en particulier chez les patients NMOSD AQP4Ab positifs. L’utilisation de ce médicament l’a porté en première ligne des traitements de fond du NMOSD, a fortiori avec un profil de sécurité favorable. Les effets secondaires sont avant tout représentés par des réactions aux infusions et le développement d’une diminution du taux de globules blancs avec parfois une diminution des taux d’immunoglobulines (anticorps qui nous défendent vis-à-vis des infections). L’utilisation régulière du Rituximab en Belgique n’a été possible que depuis un peu plus de 2 ans.
Le Mycophénolate Mofétil (MMF)
Le MMF a été initialement utilisé dans le traitement antirejet chez des patients greffés. Cette molécule exerce des effets d’inhibition de la prolifération cellulaire, en particulier sur les lymphocytes mais aussi les macrophages et les cellules dendritiques. Le MMF diminue la production des anticorps. La dose habituelle est de 1500 à 3000 mg par jour en deux prises. L’effet favorable du MMF dans le NMOSD n’a été évalué que de manière rétrospective avec des résultats assez variables mais globalement positifs. Le médicament nécessite souvent plusieurs mois pour atteindre sa pleine efficacité et son association avec des corticoïdes est courante, surtout au début du traitement. Des taux de 15 à 25 % d’intolérance sont rapportés, essentiellement sous la forme de troubles digestifs ou d’infections.
L’Azathioprine (AZA)
Ce médicament a été aussi initialement utilisé comme antirejet à partir des années 1960. Il s’agit d’une pro-drogue qui est modifiée par l’organisme en molécules actives. Ces dernières exercent un effet d’inhibition de prolifération lymphocytaire avec une réduction de la synthèse des anticorps. Les doses varient entre 1 et 4 mg/kg/jour. Des problèmes d’intolérance digestive sont relativement fréquents (4 %). Des infections sévères et une toxicité hépatique sont également rapportés dans 2 % des cas. Le maintien des corticoïdes pendant 3 à 6 mois est nécessaire pour atteindre la pleine efficacité de l’AZA. Les données scientifiques sont assez conflictuelles en ce qui concerne l’efficacité relative de l’AZA par rapport au MMF ou au Rituximab.
La Mitoxantrone (MITOX)
Ce médicament est un produit de chimiothérapie initialement utilisé dans le traitement de certains cancers (surtout le cancer du sein). Les effets immunologiques de cette molécule sont larges : inhibition de la fonction des lymphocytes B (dont la sécrétion d’anticorps), inhibition de certains types de lymphocytes T et réduction de la production de cytokines (molécules de communication entre les acteurs de la réaction immunitaires) pro-inflammatoires. Plusieurs publications scientifiques rapportent des effets favorables importants dans le NMOSD mais les séries sont généralement assez limitées et plusieurs facteurs confondants rendent délicate l’interprétation des résultats. Le risque d’induction d’une leucémie est estimé à 1 % à dix ans, ce qui limite nettement l’utilisation de ce médicament actuellement.
Le Cyclophosphamide (CYC)
Le CYC est aussi une pro-drogue qui interfère avec la division cellulaire. L’effet immunosuppresseur est large, tant sur les lymphocytes B que les lymphocytes T. Il exerce aussi une action de modification de la balance immune en faveur d’une régulation. Les premières utilisation de ce médicament dans le NMOSD viennent du traitement de patients qui présentaient plusieurs affections auto-immunes en association avec le NMOSD (lupus, maladie de Sjögren). Le nombre de cas rapportés est limité. La dose orale usuelle est de 50 mg par jour.
Les médicaments de dernière génération et en cours de mise au point
Inebilizumab (MEDI-551)
Il s’agit d’un anticorps monoclonal cytotoxique humanisé dirigé contre le CD19, de type IgG1 kappa. Son action toxique sur les lymphocytes est dépendante de l’activation du système du complément et est théoriquement plus large que celle produite par le Rituximab. En 2019, une étude de phase 2 a été publiée dans un importante revue scientifique (The Lancet). Cette étude a regroupé 230 patients traités par la molécule active ou un placebo. Le recrutement dans la phase double aveugle a été précocement arrêté compte tenu de la rapide démonstration d’un effet favorable important du médicament avec un bon profil de tolérance.
Satralizumab (SA237)
Il s’agit aussi d’un anticorps monoclonal humanisé (IgG2) qui est dirigé contre l’interleukine-6. L’interleukine-6 est une cytokine pro-inflammatoire largement synthétisée par les lymphocytes B et T mais aussi les monocytes et les fibroblastes. Il s’agit d’une molécule très proche d’un autre anticorps monoclonal anti-IL6, le Tocilizumab, utilisé en rhumatologie. L’action concerne non seulement les cellules productrices d’anticorps mais aussi la perméabilisation de la barrière hémato-encéphalique. Une étude de phase III a été publiée dans le New England Journal of Medicine (NEJM) en novembre 2019 avec la démonstration d’un effet favorable très marqué par rapport au placebo chez 83 patients inclus, alors qu’ils recevaient par ailleurs un traitement immunosuppresseur classique.
Eculizumab
Il s’agit toujours d’une anticorps monoclonal humanisé qui, cette fois, se fixe sur la fraction terminale d’une composante du système du complément (C5). Il provoque un blocage de cette filière pro-inflammatoire. Ce médicament est déjà approuvé aux USA pour d’autres maladies auto-immunes (hémoglobuniurie paroxystique, syndrome hémolytique urémique atypique et myasthénie grave). Les résultats d’une étude de phase III contre placebo ont été publiés dans le NEJM en 2019. Ils sont très nettement favorables. Une analyse intermédiaire des données d’extension de cette étude viennent d’être publiés, en février 2021, dans une autre grande revue scientifique internationale (Annals of Neurology). Un éditorial a rapporté ces effets très positifs et a souligné l’importance d’une vaccination préalable contre le méningocoque avant l’utilisation de ce médicament qui expose les patients à un risque plus élevé vis-à-vis de cette infection grave. Des développements sont en cours pour modifier la molécule et tenter de réduire la fréquence d’administration du médicament.
Les molécules en cours de développement
L’Ublituximab est un autre anticorps monoclonal anti-CD20 dirigé contre les cellules productrices d’anticorps. Des données récentes ont été acquises en traitement complémentaire (association avec les corticoïdes) des poussées de NMO. D’autres anticorps monoclonaux anti-CD19 ou des cellules T capables de destruction des cellules productrices d’anticorps (CAR-T cells) sont en cours d’évaluation. D’autres molécules sont en phases préliminaires d développement, chacune avec différents mécanismes d’action. Parmi elles, on citera l’Aquaporumab qui est un anticorps monoclonal destiné à se fixer au même récepteur que l’anticorps pathologique (AQP4Ab) mais avec une plus haute affinité et surtout un blocage des mécanismes d’attaque de la cellule cible. Il s’agit donc d’une molécule destinée à entrer en compétition avec l’anticorps pathologique et ainsi le bloquer. La restauration de l’intégrité de la barrière hémato-encéphalique est également une voie de recherche qui comporte notamment l’utilisation du bevacizumab. Cette molécule est utilisée en cancérologie et bloque un agent qui favorise le développement de nouveaux capillaires sanguins (VEGF). Des travaux menés chez un petit nombre de patients ont montré un effet favorable comme traitement de la poussée, en association avec des corticoïdes. D’autres voies de recherche s’attachent à agir sur les polynucléaires qui sont impliqués dans la cascade inflammatoire à l’origine des lésions de NMO. Un médicament utilisé dans des pathologies respiratoires aigües sévères pourrait être intéressant par son action sur les polynucléaires neutrophiles. Un antihistaminique « simple », la cétirizine, pourrait avoir un effet favorable par son action sur les éosinophiles et limiterait le risque de rechute. Des travaux à petite échelle vont dans ce sens mais nécessitent des confirmations plus larges. Le Bortezomib est un médicament utilisé par les oncologues. Il s’agit d’un inhibiteur du protéasome qui est un système complexe de survie cellulaire. Il a été testé chez un petit nombre de patients NMO réfractaire à l’azathioprine ou au Rituximab avec des résultats intéressants.
En conclusion
Au cours des quinze dernières années, les NMOSD sont devenues une entité à part entière avec une compréhension des mécanismes physiopathologiques principaux qui sont en cause et qui diffèrent clairement de ceux de la SEP. La démonstration anticorps anormaux a permis de développer des approches pharmacologiques bien plus précises. Des efforts de coordination de la recherche clinique à l’échelon mondial ont permis d’aboutir à des études de haute valeur scientifique. Elles changent complètement le paradigme de prise en charge de cette maladie. Les années qui viennent permettront aux patients de bénéficier de nouveaux médicaments et d’une prise en charge optimisée qui leur permettra de modifier complètement le sombre pronostic fonctionnel auquel ils étaient soumis par le passé.
Dominique Dive, CHU de Liège, 21 février 2021.
Référence principale et schéma annexe : Collongues, N., et al. (2019). "Pharmacotherapy for Neuromyelitis Optica Spectrum Disorders: Current Management and Future Options." Drugs 79(2): 125-142.
La fatigue et la sclérose en plaques
- En quoi consiste cette fatigue ?
- Comment explique-t-on cette fatigue ?
- Peut-on mesurer objectivement cette fatigue ?
- Comment vivre avec cette fatigue ?
- Peut-on prévenir ou soigner la fatigue ?
- Peut-on améliorer la fatigue avec des médicaments ?
Le nombre de facteurs de risque connus pour la sclérose en plaques a doublé
Des scientifiques ont identifié 29 nouveaux facteurs de risque héréditaires pour la sclérose en plaques (SEP), ce qui porte leur total à plus de 50. Ces gènes de risque n'ont qu'une faible valeur prédictive pour les personnes atteintes de SEP ou les membres de leur famille. Leur importance réside toutefois dans le fait qu'ils permettent de mieux comprendre l'apparition de la maladie. Cette étude génétique sur la SEP, la plus grande jamais réalisée, n'aurait tout simplement pas été possible sans un vaste réseau international de chercheurs et sans la participation de milliers de personnes confrontées à cette terrible maladie.
La SEP n'est pas une maladie héréditaire, mais certaines prédispositions héréditaires entraînent un risque accru. Ainsi, la maladie est un peu plus répandue dans les mêmes familles : une personne sur cinq atteintes de SEP a un ou plusieurs membres de sa famille qui en souffrent aussi. Le premier facteur de risque héréditaire pour la SEP a été découvert dans les années 1970. Ensuite, malgré de nombreux efforts, il est apparu difficile d'enregistrer des progrès. Depuis quelques années, nous disposons toutefois des connaissances et des instruments qui se sont révélés les clés de la réussite d'études génétiques.
Une équipe internationale de chercheurs ont réuni leurs forces dans un partenariat baptisé International Multiple Sclerosis Genetics Consortium et Wellcome Trust Case Control Consortium. L'équipe était dirigée par les universités de Cambridge et d'Oxford. La Belgique a apporté sa collaboration à cette étude par le biais du laboratoire de neuro-immunologie de la K.U.Leuven. Nous avons fait état de nos conclusions le 11 août dans le célèbre magazine scientifique Nature. Cette étude est la plus grande étude génétique jamais réalisée sur la SEP. Près de 250 chercheurs de plusieurs pays ont étudié le matériel héréditaire ou ADN de 9.772 personnes atteintes de SEP et de 17.376 personnes de contrôle saines issues de 15 pays différents. Parmi eux, plus de 500 personnes atteintes de SEP ont participé à cette étude en donnant un échantillon de sang aux Hôpitaux universitaires de Leuven ou au Centre national de la sclérose en plaques de Melsbroek.
Variantes héréditaires
Dans un premier temps, nous avons confirmé le rôle de 23 facteurs de risque héréditaires déjà connus. En outre, nous avons identifié 29 nouvelles variantes héréditaires qui déterminent en partie la prédisposition pour la maladie, ce qui double le nombre total de facteurs de risque héréditaires et porte leur nombre à plus de 50. Bon nombre de ces facteurs de risque sont des variations de gènes qui jouent un rôle essentiel dans le système immunitaire, par exemple dans la fonction des lymphocytes T (un type de globules blancs responsable de l'immunité contre des bactéries ou des virus, mais qui joue aussi un rôle dans la réaction contre son propre corps dans des maladies autoimmunes) et l'activation d'interleukines (protéines messagères entre différents types de cellules immunitaires). Il est frappant de constater qu'un tiers des facteurs de risque de la SEP avait déjà un rôle connu dans d'autres maladies auto-immunes (par ex., la maladie de Crohn et le diabète de type 1). Ce constat démontre que les mêmes mécanismes jouent un rôle dans plus d'un type de maladies auto-immunes.
D'anciennes études suggéraient un lien entre le déficit en vitamine D et un risque accru de SEP (voir Bulletin d'information du 29 mai 2011). Dans la présente étude, outre les facteurs de risque qui jouent un rôle direct dans le système immunitaire, nous avons identifié deux facteurs de risque qui participent à la transformation de la vitamine D. Ces découvertes nous permettent de mieux comprendre la relation entre des facteurs héréditaires et environnementaux, qui jouent tous les deux un rôle dans la SEP.
La mesure dans laquelle les facteurs de risque héréditaires augmentent le risque d'une personne de contracter la SEP est trop faible pour pouvoir être utilisée pour prévoir qui développera la maladie ou non. Il ne s'agit en l'occurrence pas de «fautes» rares dans le matériel génétique comme nous pouvons le voir avec des maladies héréditaires comme certains cancers. Il s'agit bien d'une variation normale entre personnes.
Cette variation normale détermine entre autres diverses caractéristiques de personnes, telles que la couleur des cheveux et des yeux, la prédisposition à l'obésité ou la prédisposition à des maladies telles que la SEP. Une personne présentant de nombreux facteurs de risque héréditaires pour la SEP a un peu plus de probabilité de développer la maladie qu'une personne qui présente moins de facteurs de risque héréditaires.
Nouvelles perspectives de recherche
La découverte des bases de la prédisposition héréditaire pour une maladie fournit des éléments fiables permettant de comprendre les mécanismes de cette maladie. Ainsi, l'étude révèle les cellules et les protéines du système immunitaire qui sont cruciaux dans le développement de la maladie. Ces connaissances sont importantes pour mieux comprendre la SEP et pour en améliorer le traitement. Preuve en est le fait que deux des facteurs de risque héréditaires sont la cible de traitements existants. Les autres facteurs de risque peuvent révéler de nouvelles cibles pour des traitements futurs.
Le progrès majeur réalisé grâce à cette étude ouvre de nouvelles perspectives dans la recherche sur la SEP. Nous devons à présent étudier de quelle manière ces facteurs de risque héréditaires, seuls ou en combinaison, exercent précisément leur influence sur la prédisposition à la maladie et comment nous pouvons intervenir à ce niveau pour traiter le mieux possible la SEP.
Prof A. Goris
Laboratoire de neuro-immunologie,
Département Sciences neurologiques, KULeuven
Prof B. Dubois
Service Neurologie, Universitaire Ziekenhuizen Leuven
Nouvelles perspectives thérapeutiques dans la SEP
La sclérose en plaques est une maladie qui « joue » un double jeu. Le premier est celui d'une inflammation intermittente, qui se présente sous forme de poussées, ou sous forme de plaques « actives » en résonance magnétique, sans que l'on s'en rende compte nécessairement (une plaque active est une plaque en état inflammatoire, captant le produit de contraste injecté par voie intra-veineuse). Le deuxième est celui d'une lente dégénérescence des fibres nerveuses privées de leurs gaines de myéline, ou sectionnées le long de leur trajet par l'inflammation initiale.
Les nouveaux traitements qui se profilent à l'horizon sont essentiellement anti-inflammatoires, et ne protègent pas de la neuro-dégénérescence provenant de lésions déjà établies. Ils ne réparent pas non plus la gaine de myéline. Ils stabiliseront la maladie mieux qu'aujourd'hui. S'il y a amélioration, ce sera grâce à la « bonne nature » qui est capable de remyéliniser certaines plaques si l'inflammation est supprimée ; hélas, ces possibilités naturelles de réparation sont le plus souvent insuffisantes et dépassées par l'extension des lésions. Jusqu'à présent, les essais thérapeutiques dans les formes d'emblée progressives sans poussées, ou devenues avec le temps progressives sans poussées, n'ont pas été couronnés de succès.
A l'heure actuelle, nous disposons dans les formes avec poussées et rémissions (soit complètes, soit partielles) de 4 types d'interférons bêta (Avonex, Bêtaféron, Extavia et Rebif) et d'un 5ème produit tout à fait différent, le copolymère I (Copaxone). Ces 5 produits, qui représentent la 1ère ligne de traitement, s'administrent par injections, soit sous-cutanées, soit intra-musculaires. Ils ont chacun leurs inconvénients et leurs avantages. Ils ont globalement la même efficacité quand on compare des groupes de patients traités par ces différents produits, mais individuellement, il existe des « bons », des « moyens » et des « non »-répondeurs à chaque type de traitement. Un « bon » répondeur verra la fréquence de ses poussées diminuer de moitié, les poussées résiduelles auront en moyenne une sévérité diminuée de moitié, les lésions en résonance magnétique n'augmenteront que de quelques pourcents sur 5 ans (contre 27% en l'absence de traitement). A moyen (5 ans) et long terme (20 ans), ces produits sont extrêmement sûrs, sans effets secondaires graves.
En cas d'échec de ces produits malgré un traitement bien suivi (forte poussée, aggravation de l'imagerie cérébrale, plaques actives nombreuses), nous avons recours au Tysabri, un produit biologique injecté une fois toutes les 4 semaines par voie intra-veineuse, qui bloque le passage des lymphocytes sanguins (cellules immunitaires principales) du sang vers le cerveau ou la moelle. C'est actuellement le traitement le plus efficace des formes très actives, caractérisées par la répétition de poussées très fréquentes. Malheureusement, il existe un risque, faible mais réel, de développement d'une encéphalite provoquée par un virus qui envahit le cerveau et qui tue spécifiquement les cellules fabriquant la gaine de myéline. Il n'existe pas de médicament contre ce virus. Nous donnons le Tysabri quand nous sommes convaincus que les bénéfices l'emportent largement sur le risque d'encéphalite. Enfin, nous utilisons la Novantrone par voie intra-veineuse, malgré sa toxicité (insuffisance cardiaque, leucémie...) chez les personnes avec poussées et aggravation progressive de la maladie associée à la persistance de poussées et de plaques actives en imagerie.
Les nouveaux médicaments vont se situer au même niveau que les 5 premiers cités, ou entre ces 5 produits et le Tysabri : ils seront aussi efficaces, ou plus efficaces que l'actuelle 1ère ligne, et plus confortables (prise orale). Leurs effets secondaires à cours et moyen terme sont cependant inconnus. Il n'y aura pas de motif de changer le traitement d'une personne qui supporte bien les injections des produits actuels, et qui est en rémission tant sur le plan clinique que radiologique (imagerie du cerveau et de la moelle).
- Le Gilenya ® (Fingolimod), une gélule par jour, agit sur le trafic des lymphocytes et les re-dirige vers les ganglions pour les empêcher de traverser la barrière entre le sang et le cerveau. Les résultats publiés sont très positifs. Il a été accepté par l'administration américaine des médicaments sur le même plan que les 5 médicaments actuels de 1ère ligne, et par l'agence européenne, en cas d'échec de la 1ère ligne, sur le même plan que le Tysabri. Il doit être maintenant soumis aux autorités de la Santé de chaque pays européen, et nous connaissons la lenteur des procédures belges. Il serait de toute façon inadmissible que les indications belges soient plus restrictives que les conditions européennes, comme ce fut le cas initialement pour le Tysabri.
- La cladribine est un immunosuppresseur par voie orale, à longue durée d'action, à ne prendre que quelques jours par an. L'agence européenne n'a pas accepté ce produit dans le cadre de la SEP, par crainte de risques à long terme. L'administration américaine n'a pas encore donné son avis. Les résultats publiés sont aussi très positifs, et des études complémentaires et prolongées sont en cours.
- Le fumarate (BG12), le teriflunomide, le laquinimod sont d'autres produits par voie orale, testés dans la SEP, avec une efficacité au moins similaire à la 1ère ligne actuelle.
Enfin, d'autres médications sont en cours de tests, dont l'action pourrait être plus radicale : tuer dans l'œuf la maladie dès sa première manifestation, et « re-formater » le système immunitaire. Il s'agit de l'Alentuzumab (Campath) et des anticorps anti-lymphocytes B comme le rituximab et l'ocrélizumab.
La conclusion est claire : dans 5 ans, le traitement de la SEP se sera profondément modifié mais il s'agira pour chaque patient(e) de faire la balance entre risques, bénéfices, qualité de vie, projets personnels (fonder une famille par exemple). Et dans 5 ans, il y aura encore beaucoup de recherches à réaliser, pour arrêter les formes progressives de la maladie et si possible, réparer les lésions déjà présentes.
Professeur C. SINDIC
Une nouvelle ère pour le traitement de la SEP ?
Les 12, 13 et 14 novembre 2009, s'est tenue à Lisbonne la réunion Charcot qui réunissait de nombreux spécialistes de la SEP pour faire le point sur les progrès scientifiques réalisés dans cette maladie et sur les espoirs raisonnables que ces progrès suscitent en matière de thérapeutiques nouvelles.
Il est évidemment impossible de résumer, même de manière partielle, tout ce qui a été dit d'intéressant au cours de cette réunion, mais on peut cependant dégager les 2 notions suivantes :
- Première notion : La recherche fondamentale en matière se SEP se traduit par un énorme apport d'informations nouvelles relatives aux mécanismes de la maladie en termes de biologie moléculaire. Toutes ces informations ne se traduiront pas rapidement en nouveaux traitements de la maladie, mais elles constituent certainement des pistes extrêmement intéressantes et prometteuses pour concevoir ces nouveaux traitements et les mettre au point à échéance moyenne.
- Deuxième notion : Des traitements nouveaux sont en cours d'investigation chez des patients atteints de SEP. Plusieurs molécules sont en phase III (pré enregistrement), et des résultats définitifs sont attendus dans les 3 prochaines années. Même si toutes les molécules ne deviendront pas nécessairement des traitements de la SEP, on peut raisonnablement espérer l'arrivée prochaine sur le marché de quelques produits doués d'activités significatives et surtout de produits pouvant être administrés par voie orale.
Molécules nouvelles
Les molécules dont on parle le plus sont tout naturellement celles dont l'investigation est la plus avancée. Il y a ainsi une série de produits qui sont administrés depuis de nombreux mois à des cohortes de patients atteints de SEP. Ces patients font l'objet d'une surveillance très attentive qui permet d'apprécier leur réponse à ces traitements nouveaux en termes cliniques (nombre de poussées et évolution du handicap) et en termes d'examens spéciaux (examens répétés de résonnance magnétique).
D'une manière pratique, les molécules les plus souvent mentionnées sont les suivantes :
- La CLADRIBINE
- Le FINGOLIMOD
- Le LAQUINIMOD
- Le FUMARATE
- L'ALEMTUZUMAB
- La PIXANTRONE
- Une STATINE...
(Liste non exhaustive)
Dans le présent document, nous donnerons quelques détails concernant la Cladribine, le Fingolimod et le Laquinimod dont l'étude est déjà bien avancée et nous attendrons que l'étude des molécules suivantes soit plus avancée pour avoir matière à des explications détaillées qu'il serait difficile de fournir aujourd'hui.
La CLADRIBINE
La Cladribine est une substance utilisée depuis des années dans le traitement de la leucémie. Après une administration orale de brève durée, elle diminue de manière importante et prolongée le nombre de lymphocytes T circulants. Chez des patients avec SEP à poussées et rémissions, cette chute de globules blancs s'accompagne d'une réduction marquée du nombre de poussées et de l'apparition de lésions nouvelles observées par résonance magnétique. Cet effet bénéfique est au moins égal, sinon supérieur à celui d'un Interféron, mais ceci reste à démontrer dans une étude comparative.
L'intérêt de la Cladribine est son mécanisme d'action qui est différent de celui des Interférons et de l'acétate de glatiramer, ce qui permet de croire que ce produit pourrait être efficace chez les patients qui ne répondent pas favorablement aux autres traitements.
Le FINGOLIMOD
Le Fingolimod est un analogue de la Sphingosine. Sa liaison au récepteur pour la Sphingosine situé à la surface du lymphocyte T a pour résultat de séquestrer les lymphocytes T agressifs dans les ganglions lymphatiques, ce qui se traduit, chez les malades atteints de la forme à rechutes et à rémissions de la SEP par une diminution importante du nombre de poussées et de l'apparition de lésions nouvelles observées par résonance magnétique. Dans une étude effectuée sur plus de mille patients traités pendant une durée d'un an, le Fingolimod s'est avéré plus efficace qu'un Interféron.
Comme pour la Cladribine, l'intérêt de ce produit est son mécanisme d'action original qui laisse prévoir une efficacité chez des patients qui ne répondent pas aux traitements traditionnels. Il s'agit également d'un produit actif par voie orale.
Le LAQUINIMOD
Le Laquinimod est une petite molécule de synthèse qui modifie la proportion des lymphocytes Th1 par rapport aux lymphocytes Th2. Ceci conduit à une augmentation relative de ces derniers, et à une augmentation relative des cytokines produites par ce type de lymphocytes.
Or, on sait que l'inflammation qui caractérise les lésions cérébrales des patients atteints de SEP dépend intimement de la présence de certaines cytokines qui sont soit pro-inflammatoires, soit anti-inflammatoires. (Pro inflammatoires : les cytokines produites par les Th1, et anti-inflammatoires, celles produites par les Th2). Administré par la bouche à des patients atteints de SEP, le Laquinimod diminue le nombre de poussées et cette diminution s'accompagne d'une incidence diminuée de lésions cérébrales mises en évidence par la résonance magnétique.
Il s'agit ici aussi d'un mode d'action original totalement différent des thérapeutiques existantes.
Effets indésirables des nouvelles molécules
Comme tout le monde le sait, un point crucial pour l'acceptation de nouvelles molécules par les commissions d'enregistrement est la démonstration de leur bonne tolérance. Non seulement la molécule nouvelle doit démontrer son efficacité, mais elle doit aussi démontrer qu'elle n'est ni dangereuse, ni toxique.
Qu'en est-il en ce qui concerne les 3 molécules mentionnées ci-dessus ? Il y a une chose importante qu'il convient d'avoir à l'esprit, et c'est la suivante : si la démonstration de l'efficacité thérapeutique d'une molécule dans la SEP est une démarche difficile et coûteuse pour les laboratoires, la démonstration de la bonne tolérance est une démarche bien plus exigeante encore. Il faut quelques centaines de patients traités pendant 2 ans pour s'assurer de l'efficacité d'un produit, et il faut des milliers de patients traités pendant des périodes beaucoup plus longues pour s'assurer de l'innocuité d'un produit.
En pratique, cela signifie que bien des médicaments nouveaux sont mis sur le marché avant que l'on puisse totalement rassurer les malades et les médecins quant à l'absence d'effets indésirables au long terme. Cette réalité impose naturellement de procéder à un suivi très attentif des patients auxquels on administre un nouveau produit, surtout s'il s'agit d'un produit qui exerce une action sur le système immunitaire. Avec ce type de produits, on doit s'attendre en effet à voir survenir certaines complications infectieuses et parmi celles-ci, la leucoencéphalite multifocale progressive (infection cérébrale virale particulièrement grave.) Un suivi adéquat permettra de déceler plus rapidement ces effets indésirables éventuels et permettra ainsi d'en diminuer les conséquences.
En conclusion
Le traitement de la SEP va bientôt s'enrichir de molécules nouvelles douées de mécanismes d'action variés. On disposera alors d'un véritable arsenal thérapeutique, et on pourra sans doute envisager de combiner différents produits avec l'espoir d'obtenir un meilleur contrôle de la maladie et de sa progression. Cela va sans doute modifier considérablement la prise en charge thérapeutique des patients atteints de SEP. Cette perspective est évidemment réjouissante, et il faut reconnaître qu'un vent d'optimisme soufflait sur la réunion Charcot à Lisbonne.
Cela ne nous fait pas oublier les difficultés quotidiennes vécues par de nombreuses personnes atteintes de SEP. Cette vie de tous les jours se présente souvent pour elles comme un véritable défi. Les médecins ne l'oublient pas, même quand ils annoncent la bonne nouvelle. Il est vrai que les malades ont souvent cette impression que les progrès de la médecine ne sont pas rapides : pour eux, et on peut le comprendre, les choses ne vont jamais assez vite.
Dr.J.P. Rihoux
Février 2010
Sclérose en plaques et vaccination
Le Conseil Médical de la Ligue Nationale de la SEP a étudié les questions soulevées par les vaccinations chez les personnes atteintes de sclérose en plaques lors de sa réunion du 6 octobre 2009, dans le contexte des risques d'épidémie de grippe par le nouveau virus A/H1N1. Il tient dès lors à préciser les éléments suivants :
- Aucun vaccin n'a été démontré néfaste chez les personnes atteintes de sclérose en plaques. Aucune vaccination n'est à l'origine d'un déclenchement de sclérose en plaques, d'une poussée de sclérose en plaques ou de son aggravation. Le vaccin anti-tétanique pourrait même avoir un léger effet protecteur.
- Le vaccin contre la grippe saisonnière a été très spécifiquement étudié. Il est parfaitement bien toléré et n'induit pas de poussées de SEP, ni de nouvelles lésions détectables en résonance magnétique cérébrale. En cas d'épidémie de grippe, les patients vaccinés font significativement moins de poussées que les patients non-vaccinés.
- Les personnes atteintes de SEP ne sont pas plus vulnérables, ni plus menacées par le nouveau virus A/H1N1, que la population générale.
- Il n'y a aucun élément faisant suspecter que le vaccin contre ce nouveau virus de la grippe soit moins bien toléré, ou plus dangereux que le vaccin habituel contre la grippe saisonnière.
- Les personnes atteintes de SEP, pour qui leur médecin traitant ou leur neurologue recommande habituellement le vaccin anti-grippe, peuvent dès lors bénéficier aussi de la vaccination contre le nouveau virus A/H1N1, en respectant un intervalle de 3 à 4 semaines entre les deux vaccinations.
- La vaccination anti-influenza, tant pour la grippe saisonnière que pour le nouveau virus A/H1N1, peut être pratiquée sans danger chez les patients traités par interférons bêta (Avonex, Bétaféron, Rebif), par Copaxone et par Tysabri, ainsi que par Novantrone, Imuran ou Ledertrexate.
Pour ces trois derniers produits, elle pourrait éventuellement être un peu moins efficace. De toute façon, elle n'assure jamais une protection totale de 100%.
Octobre 2009
Les médecines parallèles dans la SEP: une alternative valable ?
Le recours aux médecines parallèles dans la sclérose en plaques est fréquent mais reste controversé. Comme ces traitements alternatifs sont utilisés seuls, ou en combinaison avec des traitements conventionnels, il est intéressant de s'interroger sur leur signification et sur leur place réelle dans le traitement global de la sclérose en plaques.
Que sont les traitements alternatifs ?
Il s'agit de traitements réputés efficaces dans certaines maladies ou contre certains symptômes particuliers, sans que leur efficacité ait été réellement prouvée et sans la démonstration que leurs éventuels effets secondaires sont acceptables.
Dans la sclérose en plaques, on peut diviser schématiquement les traitements alternatifs en deux groupes : ceux qui pourraient agir sur l'évolution de la maladie et ceux qui pourraient être actifs sur certains symptômes bien définis.
Dans tous les domaines de la médecine, les nouveaux médicaments font l'objet d'études cliniques pendant plusieurs années et leur efficacité clinique doit être démontrée de manière convaincante pour une indication précise, avec démonstration que les effets secondaires sont limités et acceptables. Pour les traitements alternatifs, la démonstration de leur efficacité est beaucoup plus ténue : ils sont utilisés sur base de quelques expériences positives, sans données précises quant à leur mode d'action, leur efficacité et leurs effets secondaires.
Fréquence d'utilisation des médecines parallèles
Les traitements alternatifs, dérivés des médecines parallèles sont surtout utilisés dans des affections pour lesquelles il n'existe pas encore un traitement complètement efficace. C'est donc pour ces maladies, surtout quand elles sont chroniques et incurables, que l'on aura recours rapidement à des traitements dont l'efficacité n'est cependant pas prouvée. Un grand nombre de personnes souffrant de sclérose en plaques (50 à 60 %) ont donc recours durant l'évolution de leur maladie à de tels traitements alternatifs, soit de manière épisodique, soit parfois pendant de longues périodes.
Dans une étude allemande portant sur 254 personnes souffrant de sclérose en plaques, les personnes ayant recours à des traitement alternatifs étaient âgés en moyenne de 44 ans avec une durée moyenne de la maladie de 8 ans. Leur échelle d'invalidité, mesurée par l'EDSS (Expanded Disability Scale Score) était de 4, ce qui signifie qu'ils étaient encore capables de marcher 500 mètres sans avoir recours à une aide. Le sexe, l'éducation, les croyances religieuses ne jouaient aucun rôle dans ce recours aux médecines parallèles.
En moyenne, les patients ayant recours à des traitements alternatifs avaient une maladie d'une durée plus longue, un handicap plus sévère, et étaient plus âgés que les personnes n'utilisant pas de tels traitements.
Quels sont les traitement alternatifs existant dans la sclérose en plaques ?
Il est impossible d'établir une liste complète de tous les traitements alternatifs qui ont été proposés dans la sclérose en plaques. En ce qui concerne les traitements recommandés pour modifier l'évolution à long terme de la maladie, il existe des régimes, par exemple riches en acide gras polyinsaturés ou en antioxydants. D'autres traitements sont surtout recommandés pour diminuer certains symptômes de la maladie : immersion en eau froide, acupuncture, yoga... Enfin, il existe des médications alternatives qui pourraient agir à la fois sur l'évolution de la maladie et sur ses symptômes : le cannabis, la vitamine D, le gingko biloba. Certains de ces traitements paraissent prometteurs, d'autres sont nocifs, mais dans tous les cas leur efficacité est insuffisamment démontrée.
Les traitements alternatifs sont-ils utiles ?
Certains de ces traitements alternatifs pourraient certainement être utiles, mais hélas leurs effets positifs n'ont jamais été prouvés. Pour la plupart de ces traitements, il n'y a jamais eu d'études réalisées; pour d'autres, les études réalisées n'ont apporté que des preuves insuffisantes d'une efficacité thérapeutique.
Les traitements alternatifs sont-ils nocifs ?
Comme non seulement les effets positifs, mais aussi les effets secondaires possibles des traitements alternatifs n'ont pas été étudiés suffisamment, il n'est pas exclu que certaines thérapeutiques parallèles soient nuisibles. En plus d'un effet nuisible direct, il existe aussi le danger que l'usage incontrôlé de ces traitements alternatifs puisse devenir par lui même dangereux. Par ailleurs, le coût de ces traitements est souvent trop élevé par rapport à l'effet favorable que l'on pourrait en attendre.
Conclusion
L'utilisation de médications relevant de médecines parallèles dans le traitement de la sclérose en plaques est largement répandue. Il n'est pas exclu que certaines de ces médications puissent effectivement moduler l'évolution de la maladie, ou soulager certains symptômes particuliers. Il serait tout à fait raisonnable que ces traitements alternatifs fassent l'objet des mêmes études et doivent répondre aux mêmes critères d'efficacité que les traitements médicaux classiques avant d'être utilisés souvent de manière incontrôlée.
Texte : BD, Traduction : CS, Mars 2005
Le stress et la sclérose en plaques
Voilà déjà plus de cent ans que Charcot déclarait que le stress psychologique pouvait induire des poussées de sclérose en plaques. La plupart des personnes souffrant de sclérose en plaques ressentent que des événements stressants peuvent jouer un rôle dans l'apparition de crises de cette maladie. De nombreuses études ont été publiées à ce sujet ces dernières années. Aussi a-t-on fait appel au scanner du cerveau par résonance magnétique.
Dans une étude récente sur les liens entre stress, traumatismes et sclérose en plaques, les données de 20 études publiées précédemment (période de 1965 à 2003) ont été analysées de manière critique. Après un examen approfondi de la qualité de ces études, 14 d'entre elles ont satisfait aux critères permettant de regrouper leurs données (méta-analyse). Dans 13 parmi les 14 études, on découvrit un effet similaire du stress sur les poussées de sclérose en plaques : la probabilité de crise augmente à la suite d'un événement stressant de la vie.
L'intensité du phénomène est modérée mais cliniquement significative. Une seule étude semble mettre le phénomène inverse en évidence : pendant la première guerre du Golfe, les attaques à la roquette sur Tel Aviv ont provoqué moins de poussées de sclérose en plaques! L'origine de cette discordance n'est pas claire.
(Source : British Medical Journal 2004 ; 328 ; 731-735)
Dans une deuxième étude, on a analysé l'état de santé de parents danois qui avaient perdu un enfant de moins de 18 ans. Cet état de santé fut comparé à celui d'un groupe de parents n'ayant pas perdu d'enfant. Le risque de développer la sclérose en plaques s'avère 50% plus élevé chez les parents ayant perdu un enfant que chez les autres.
C'est seulement après 8 ans que ce lien devient clair. Si la perte d'un enfant arrive de manière inattendue, le risque de voir se développer la sclérose en plaques est doublé.
Ces résultats suggèrent qu'un événement extrêmement stressant comme la perte d'un enfant augmente le risque de sclérose en plaques.
(Source : Neurology 2004; 62 ; 726-9)
Ces résultats d'études ouvrent des possibilités de recherche sur les conséquences psychologiques, neuro-hormonales et immunologiques d'événements stressants sur la sclérose en plaques.
Juin 2004, MBD - Traduction PS
La SEP et les hypocholestérolémiants de la famille des statines
Les statines, une famille de médicaments qui réduisent le taux de cholestérol dans le sang, ont des propriétés immuno-modulatrices connues depuis longtemps.
Récemment, il a été montré que ces médicaments peuvent retarder le début de l'encéphalomyélite auto-immune expérimentale (EAE), un modèle animal de sclérose en plaques, et diminuer la gravité de la maladie.Lors de tests sur des globules blancs humains, la synthèse de molécules inflammatoires a été fortement diminuée.
Une première petite étude chez des patients souffrant de sclérose en plaques avec poussées, a donné des résultats intéressants. Avec une forte dose de simvastatine (80 mg), on constate une diminution du nombre de lésions actives à l'examen par IRM.
Des études cliniques plus importantes sont nécessaires pour déterminer l'efficacité et la sûreté de ces produits dans le cadre de la sclérose en plaques.
Source : The Lancet 2004 ; 363 :1607-8
Texte révisé : MBD - juin 2004
Traduction : CS
Exposition au soleil entre 6 et 15 ans et risque de développer une SEP
A propos de l'article de I. van der Mei et collaborateurs (British Medical Journal, 9 août 2003, vol 327 : pages 316-320).
Le SEP se déclare par un concours d'influences provenant de l'environnement d'une part, et de facteurs héréditaires de susceptibilité, d'autre part. Différents gènes (porteurs de nos caractéristiques héréditaires) semblent jouer un rôle dans le risque de développer une SEP, mais seulement quelques-uns sont connus. Les personnes porteuses d'un ou plusieurs de ces gènes, et de ce fait susceptibles de souffrir de la SEP, ne développeront la maladie que si elles sont soumises à certaines influences de l'environnement qui provoquent la SEP.
Il pourrait s'agir de certaines infections virales, de produits toxiques, de carences dans l'alimentation ou d'autres facteurs encore inconnus. C'est pourquoi la découverte d'Ingrid van der Mei, qu'un manque d'ensoleillement peut jouer un rôle dans l'origine de la SEP, est intéressante même si nous devons ajouter que d'autres chercheurs avaient déjà des indications allant dans la même voie.
Ingrid van der Mei et ses collaborateurs ont questionné en Tasmanie, une île proche de l'Australie, 136 personnes atteintes de SEP et 272 personnes en bonne santé, de même âge et de même sexe, sur leurs modes de vie et les événements vécus dans leur passé. Les questions portaient surtout sur l'exposition au soleil au fil des années (moins d'une heure par jour, 1 à 2 heures, 2 à 3 heures, 3 à 4 heures, plus de 4 heures). Une exposition faible correspondait à une moyenne journalière en dessous de 2-3 heures, une exposition élevée à une moyenne journalière au-dessus de 2-3 heures. Les données des patients SEP ont été ensuite comparées à celles des personnes interrogées n'ayant pas la SEP.
La comparaison a démontré qu'une exposition élevée au soleil, durant la période de 6 à 15 ans, diminuait de plus de la moitié le risque de développer plus tard une SEP. L'ensoleillement durant les mois d'hiver semblait avoir le plus d'effet préventif. Il est intéressant de noter qu'à un âge plus avancé, et plus précisément durant les 10 ans précédant le début de la SEP, l'exposition au soleil n'avait plus d'influence sur le risque de développer une SEP.
Les résultats de l'enquête sont confortés par des constatations faites dans une autre partie de l'étude : les personnes atteintes de SEP avaient moins de dommages cutanés provoqués par le soleil que les autres personnes de l'étude non atteintes de SEP.
Cette étude a été élaborée très soigneusement et paraît très convaincante mais le dernier mot n'a naturellement pas encore été dit. Des personnes critiques ne manqueront pas de faire remarquer qu'on peut mettre en doute la fiabilité de la mémoire sur le degré d'exposition au soleil durant sa jeunesse et son adolescence. Essayez vous même de vous le rappeler. A ceci on peut répondre que cette étude n'est pas unique et que des résultats d'études antérieures suggéraient déjà l'influence de l'ensoleillement. Ces études auront pour conséquence que l'on va intensifier les recherches portant sur l'effet des rayons UV sur l'immunité, via peut-être la vitamine D et sa forme active, la 1,25 dihydroxy-cholecalciferol.
Bien évidemment, exposer les enfants à plus de soleil n'a pas seulement un effet protecteur possible contre la SEP, mais pourrait avoir aussi un effet nocif par un risque plus élevé de cancer de la peau. Personne n'osera conseiller de s'exposer plus au soleil étant donné qu'il n'est pas possible actuellement de dire si les avantages l'emportent sur les inconvénients.
Il est aussi intéressant de noter que l'étude de van der Mei a uniquement examiné l'importance de l'ensoleillement dans la prévention de la SEP. Que le soleil ait une influence sur l'évolution de la maladie chez les personnes déjà atteintes de SEP n'a jamais encore été examiné.
On peut aussi rapprocher les observations rapportées dans cet article avec celles faites lors d'études sur les migrants, montrant que le risque de développer une SEP était acquis vers l'âge de 15 ans, et aussi avec la prévalence de la maladie dans l'hémisphère nord, plus élevée dans les pays du Nord que dans ceux du Sud, plus ensoleillés.
HC - Janvier 2004
Carte génétique de la SEP
La carte génétique de la SEP montre le rôle des cellules immunitaires sanguines et cérébrales dans l'apparition de la SEP
Cannabis et sclérose en plaques
Depuis plusieurs années déjà, le cannabis médicinal est utilisé à des fins thérapeutiques dans le traitement de la spasticité et de la douleur, notamment chez des personnes atteintes de sclérose en plaques. Cette utilisation occupe de plus en plus le devant de la scène vu l'enregistrement de Sativex® dans plusieurs pays européens. Sativex® n'est toutefois pas encore disponible en Belgique à l'heure actuelle.
Sativex® est une teinture de la plante de cannabis qui est vaporisée par voie orale. Chaque vaporisation contient une dose standardisée de 2,7 mg de tétrahydrocannabinol (THC) et de 2,5 mg de cannabidiol (CBD). Sativex® a été enregistré après la publication de quelques études mettant en avant certains arguments en faveur d'une réduction de la spasticité. L'amélioration sensible de la spasticité ressort principalement de l'évaluation subjective du traitement, tandis que la mesure objective de la spasticité ne fait souvent pas apparaître de différence significative (1, 2). L'étude qui a mené à l'enregistrement de Sativex® a démontré un effet considérable sur la spasticité mais uniquement au sein du groupe de 42 % des répondants sélectionnés au préalable (3). L'effet d'une autre forme de cannabis thérapeutique, à savoir les capsules de tétrahydrocannabinol, sur des aspects subjectifs de la spasticité comme leur gravité, les douleurs qu'elle engendre et la qualité du sommeil (4) a également été démontré récemment.
Il convient de mettre en garde contre l'utilisation du cannabis non thérapeutique, aussi bien absorbé par cigarette que par voie orale. La quantité de THC a en effet fortement augmenté. Dans les années 60, la concentration s'élevait en moyenne à 10 mg de THC par cigarette tandis qu'aujourd'hui, elle s'élève à 150 mg ou plus. Dans le cas de la cigarette, il faut compter une absorption rapide d'au moins 50 %, sans parler de ses effets cancérigènes décrits. En outre, le cannabis a un effet de longue durée compte tenu des produits de dégradation actifs et de leur récupération par les intestins. L'élimination totale d'une dose peut prendre jusqu'à 30 jours (5). La toxicité aiguë du cannabis est, certes, basse mais les vertiges, étourdissements, troubles psychiques et cognitifs sont plus fréquents. Ceux-ci apparaissent surtout chez les personnes atteintes de sclérose en plaques étant donné que des troubles cognitifs peuvent déjà se manifester chez 43 à 65 % des malades. Ces troubles cognitifs peuvent augmenter en cas de consommation de cannabis. La consommation de cannabis entraîne un ralentissement du traitement de l'information et de la vitesse psychomotrice, une diminution de la vigilance et des troubles de la mémoire vive, surtout en cas d'usage prolongé (6 à 9). Sur la base de ces données, il faut mettre en garde contre l'usage de cannabis non thérapeutique et la prudence est certainement de rigueur en cas d'usage éventuel de formes médicinales si des troubles cognitifs sont déjà présents.
Traduction du texte du Dr. D. Decoo - octobre 2012
Références :
1. Lakhan S et al - Whole plant cannabis extracts in the treatment of spasticity in multiple sclerosis: a systematic review. BMC Neurol 2009, 9:59.
2. Leussink VI et al. Symptomatic therapy in multiple sclerosis: the role of cannabinoids in treating spasticity. Ther Adv Neurol Disorders 2012; 5: 255-266.
3. Novotna A et al. A randomized, double-blind, placebo-controlled, parallel-group, enriched design study of nabiximols (Sativex ®) as add-on therapy in subjects with refractory spasticity caused by multiple sclerosis. - Eur J Neurol, 2011, 18(9): 1222-1131.
4. Zajicek JP et al. Multiple Sclerosis and extract of cannabis: results of the MUSEC trial. JNNP 2012; 83: 1125-1132.
5. Ashton C. Pharmacology and effects of cannabis: a brief review. B J Psychiatry 2001, 178:101-106.
6. Wadsworth E et.al. Cannabis use, cognitive performance and mood in a sample of workers. - J.Psychopharmacol. 2006, 20:14-23.
7. Crean R et al. An evidence-based review of acute and long-term effects of cannabis use on executive cognitive functions. - J.Addict.Med. 2011,5:1-8.
8. Rao S et al. Cognitive dysfunction in multiple sclerosis. I. Frequency, patterns, and prediction. Neurology 1991, 41:685-691.
9. Honarmand K et.al. Effects of cannabis on cognitive function in patients with multiple sclerosis. Neurology 2011, 76:1153-1160.
Relation entre le vaccin contre l'hépatite-B et la SEP
- Position du Conseil Médical de la Ligue Belge de la SEP
- Confirmation - Novembre 2004
Le Conseil Médical de la Ligue Nationale de la Sclérose en Plaques tient à rappeler très clairement l'absence de toute relation causale démontrée entre le vaccin contre l'hépatite B et le début ou l'aggravation d'une sclérose en plaques.
Une telle relation hypothétique n'a été soulevée qu'en France, à cause de la politique de vaccination massive suivie par les Autorités Sanitaires de ce pays. C'est ainsi que depuis 1994, environ 27 millions de Français ont été vaccinés contre l'hépatite B, soit près de la moitié de la population de ce pays. Cette proportion augmente à près de 66% dans la tranche d'âge comprise entre 11 et 40 ans.
Les autres pays européens ont suivi une autre politique vaccinale, consistant à vacciner les jeunes enfants, les pré-adolescents (10-12 ans) et la population adulte à risque, c'est-à-dire en contact avec des dérivés sanguins (personnel médical et para-médical).
Aucune augmentation de la fréquence de la maladie n'a été observée en France suite à cette campagne de vaccination massive. Aucune étude n'a pu montrer que les patients commençant une sclérose en plaques avaient été plus fréquemment vaccinés contre l'hépatite B que des patients de la même tranche d'âge consultant pour un autre problème neurologique. Il en ressort qu'une association temporelle entre la vaccination anti-hépatite B et le début d'une sclérose en plaques dans les semaines qui suivent est fortuite.
Le virus de l'hépatite B est 100 fois plus contagieux que celui du SIDA . Il est transmis essentiellement par contact avec le sang de « porteurs » sains ou malades, et les relations sexuelles. Le mode de transmission reste cependant inconnu dans 30% des cas. Environ un quart des porteurs sains deviendront malades et mourront de cirrhose ou de cancer du foie dans les 30 ans qui suivent la contamination. Chaque année, un million de personnes meurent de cette infection par le virus de l'hépatite B.
Nous recommandons donc que la même politique vaccinale soit appliquée aux patients atteints de sclérose en plaques et à leurs enfants et proches, que celle appliquée à l'ensemble de la population belge, à savoir :
- vaccination des enfants en bas-âge et des pré-adolescents.
- pour les enfants déjà adultes de patients atteints de sclérose en plaques, et pour les patients eux-mêmes, vaccination uniquement pour les personnes à risque (personnel médical et para-médical).
Caprivax (Goat serum)
En octobre dernier, la presse anglaise annonçait une étude en double aveugle, contrôlée par placebo avec un médicament nommé Caprivax, apparemment fabriqué avec des anticorps à base de sérum de chèvre.
Ce produit avait été crée au départ comme médicament pour le SIDA, mais il ne semblait pas très actif. En raison de ses propriétés fortement anti-inflammatoire, il est maintenant à l'essai dans le cas de SEP. Cette étude concerne des personnes atteintes de la forme secondaire progressive, encore capables de marcher (cfr Website MS society UK).
A l'heure actuelle, il n'y a pas de données disponibles dans les publications scientifiques sur le produit ou sur les effets de celui-ci dans la SEP. Le médecin responsable, le D. Barnes, n'est momentanément pas joignable par téléphone. Les experts anglais de la SEP ne sont pas au courant.
Il est indispensable de faire de plus amples recherches sur ces données avant de prendre au sérieux les effets positifs décrits (Sunday Times, 25 janvier) chez les personnes atteintes de SEP.
Février 2004
Introduction
Des études toujours plus nombreuses mettent en lumière le fait que la flore intestinale pourrait bien jouer un rôle dans notre santé, et entre autres dans le cas de la SEP ainsi que d’autres affections inflammatoires chroniques. La flore intestinale est un écosystème complexe de bactéries et d’autres micro-organismes qui communique avec le système immunitaire et le système nerveux central, et qui est également impliquée dans les interactions hormonales et le métabolisme.
Comment la flore intestinale est-elle étudiée ?
La flore intestinale est étudiée à partir d’un échantillon de selles. Le matériel génétique des micro-organismes est isolé, puis ‘lu’, ce qui permet de connaître l’identité des micro-organismes présents et ainsi de dresser le profil de la flore intestinale d’un individu. De nombreux facteurs (médicaments, durée du transit intestinal, alimentation...) peuvent influencer la composition de la flore intestinale et il est important d’en tenir compte au moment d’interpréter les résultats. Or, ce n’est pas toujours le cas. Il existe également des différences au niveau de la conservation et de la préparation des échantillons.
Résultats de la recherche
Actuellement, les recherches menées sur la flore intestinale en relation avec la SEP ne permettent pas de tirer de conclusions précises.
Dans le cadre de la recherche en Belgique, une collaboration entre l’UZ/VU Brussel, la KU Leuven et le Centre national de la Sclérose en plaques de Melsbroek a permis de mettre au jour des différences entre les caractéristiques de la flore intestinale suivant les diverses formes de la SEP chez les patients. Quelques différences ont également été relevées entre le groupe des patients SEP et le groupe de contrôle. Comme ces résultats reflètent la situation à un moment donné, on ne saurait encore évoquer un rapport de cause à effet. On n’a pas non plus encore trouvé d’indications concernant la prescription de régimes alimentaires spécifiques ou de suppléments bactériens. Par contre, ces résultats invitent à approfondir la recherche sur le rôle de la flore intestinale dans la SEP. A suivre !
Chercheurs
MB D’hooghe (Center for Neurosciences, VU Brussel, Centre national de la Sclérose en plaques, Melsbroek)
L Devolder - T Reynders - A Pauwels (chercheurs doctorants)
J Raes (Bactériologie moléculaire, KU Leuven)
A Van Remoortel (Coordinatrice d’étude, Centre national de la Sclérose en plaques, Melsbroek)
Lien vers l’article https://pubmed.ncbi.nlm.nih.gov/32162850/
Prof. M.B. D’hooghe
Juin 2021